
Valence Bond Theory of Chemical Bonding
Created | Updated Nov 20, 2006
The following entry describes the concepts of Valence Bond Theory for the structure and bonding in molecules. It is assumed that the reader has some prior knowledge of chemistry in order to fully appreciate the entire article. Some further reading on the size of molecules, electron shells and orbitals and quantum mechanics may help.
The chemical bond is of central importance to all branches of chemistry. Bonds hold atoms together to form molecules and are made and broken in the course of chemical reactions. The properties of individual molecules and bulk material are largely dependent on the nature of the chemical bonds they contain. The understanding of the underlying theories and principles is therefore key for the chemist's ability to rationalise molecular properties and successfully design and synthesise new compounds.
There are two main quantum mechanical approaches to chemical bonding: valence bond (VB) theory and molecular orbital (MO) theory. Both theories start with the same approximation; whereas the solution of the Schrodinger equation for the electron in a hydrogen atom can be solved exactly, a solution cannot be obtained for even the simplest molecule, H2+, which contains three particles: two protons and an electron. This is because all three particles are in motion relative to each other. The Born-Oppenheimer approximation is applied, which states that, since the nuclei are far heavier than the electron, they can be treated as stationary while the electrons move around them. The errors associated with this approximation are small and allow the nuclei to have a set separation in order to solve the Schrodinger equation. This allows us to then calculate the curve for potential energy as a function of inter-nuclear distance to locate the energy minimum which lies at the equilibrium separation, Re. This is the length of the bond. The first of these two approaches is valence bond theory and is considered here.
Bonding in Diatomic Molecules
The simplest molecule we can consider is molecular hydrogen, H2. To start with we have two hydrogen atoms, labelled A and B, each with an electron, labelled 1 and 2 respectively, in their 1s orbitals. Each atom can be mathematically described in terms of its electron distribution by a wave function1 expressed as ψH1s. We can generate an overall wave function describing the two electrons together by combining the wave functions of the individual atoms. This has the form:
ψ= ψH1sA(1)ψH1sB(2)
This equation describes the distribution of the two electrons with electron 1 on atom A and electron 2 on atom B. If the two hydrogen atoms are at the equilibrium bonding distance Re then the opposite arrangement is equally likely, where electron 1 is on atom B and electron 2 is on atom A. This gives the wave function:
ψ= ψH1sA(2)ψH1sB(1)
As these two arrangements of the electrons are equally likely, quantum mechanics allows us to superimpose these two wave functions. This gives us an overall wave function describing the distribution of the two electrons in the hydrogen molecule and defines the bond between the atoms.
ψ(H-H) = ψH1sA(1)ψH1sB(2) + ψH1sA(2)ψH1sB(1)
This wave function can only exist if the two electrons are paired up. This means having opposite spins in agreement with the Pauli exclusion principle2. The bond is allowed to form by the pairing of the spins of the unpaired electrons of the constituent atoms. The electron distribution results from the merging of the two spherical 1s atomic orbitals and so the wave function that describes the bond appears to be sausage-shaped. Viewed along the internuclear axis of the molecule (generally labelled the z axis by convention) this circularly symmetric wave function resembles two paired electrons in an s orbital and so is called a σ-bond, σ being the Greek letter equivalent to 's', 'sigma'. The wave function for any valence bond can be generally constructed in this way using the available singly occupied atomic orbitals on each atom present in the molecule. For a molecule A-B, we can use the atomic orbitals A and B on atoms A and B respectively to write a VB wave function of the form:
ψ(A-B) = A(1)B(2) + A(2)B(1)
To calculate the energy of a molecule, we substitute our valence bond wave function into the Schrodinger equation for a given internuclear distance, R, and solve for energy, E. When the energy is plotted against R, we see a reduction in the energy as the nuclei come together. This is because the electron on one atom is able to more freely migrate to the other atom as the nuclei become closer, and vice versa. When the internuclear separation becomes very small there is then a sharp increase in the energy. The stabilisation due to the bond formation is counteracted by the electrostatic repulsion between the two positively charged nuclei. This produces an energy minimum which is the equilibrium bond length, Re.
This approach can be applied to other more-complicated molecules where there are more valence electrons available to contribute to bonding. First we have to consider the configuration of the valence electrons for the individual atoms. For the molecule N2, we have two nitrogen atoms each with five valence electrons. For each nitrogen atom there are two electrons in the 2s orbital and one in each of the three dumbbell shaped 2p orbitals. The electron configuration for each nitrogen atom is:
N 2s22px12py12pz1
where the x, y and z labels refer to axes along which the p-orbital is aligned. Since the 2s orbital of each nitrogen atom contains two electrons that are already paired, these do not contribute to the bonding in the N2 molecule. Each p-orbital, however, contains a single unpaired electron. As the z-axis runs through two nuclei by convention, we can imagine that the 2pz orbital of one atom points at the 2pz orbital of the other atom. The 2px orbitals of each atom, perpendicular to the 2pz orbitals, are therefore parallel to each other as are the 2py orbitals. We can hence form VB wave functions by merging the matching orbitals, pairing the spins of the unpaired p-electrons. With the electrons in each 2pz orbital we form a valence bond wave function, as we did for hydrogen, that is cylindrically symmetric forming a σ-bond. In the same way that the 2pz orbitals of each atoms can merge lengthways to form a bond, the 2px orbitals of each atom and the 2py orbitals can merge sideways to pair up their electrons and form two more bonds. Viewed along the z-axis, these two VB wave functions can each be imagined to resemble two paired electrons in a p-orbital and are hence called π-bonds, π being the Greek equivalent for 'p', 'pi'. Overall we have one σ-bond and two π-bonds, giving the nitrogen molecule a triple bond and the structure :N≡N: This explains the great strength of the bond in this molecule and hence its highly inert nature.
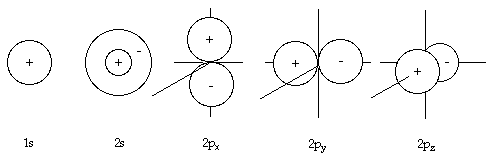{kind=link}
Polyatomic molecules.
The approach of merging singly occupied orbitals to form bonds containing paired electrons is easily extended to molecules with more than two atoms. In the water molecule, H2O, there are two hydrogen atoms, each with an unpaired electron in a 1s orbital and an oxygen atom with six valence electrons having the configuration 2s22px22py12pz1. Using both of the singly occupied p-orbitals on O, we can form two σ-bonds by pairing the electron in O 2py with the single unpaired electron of one of the H atoms and do the same with 2pz and the other hydrogen atom. As the 2py and 2pz orbitals are at right angles to each other, we would expect the two σ O-H bonds to be at 90°, however the actual bond angle is closer to 104°. Though valence bond theory seems to adequately describe chemical bonding thus far, it appears to have some deficiencies as it poorly predicts the bond angle in water and molecules such as ammonia, (NH3, predicted 90°, actually 107°). The theory gives a worse prediction than that predicted by the Valence Shell Electron Pair Repulsion (VSEPR)3 model which gives angles of about 109°. The theory also seems incapable of rationalising the tetravalent nature of carbon, whose electronic structure of 2s22px12py1 should only allow for the formation of two bonds, not four. The deficiencies in valence bond theory can be overcome by the introduction of the combined effects of promotion (excitation of an electron to a higher energy orbital) and hybridisation (the merging of the two or more atomic orbitals of the same atom in the excited state).
Promotion and Hybridisation
In valence bond theory, promotion of a paired electron is its excitation to an empty orbital at higher energy during bond formation. This energy input is worthwhile so long as it is less than the energy released through the formation of the extra bonds. In the case of carbon we have the electron configuration 2s22px12py1. Promotion of one of the paired electrons in the 2s orbital into the vacant 2pz orbital results in us having four unpaired electrons. We are now able to form four bonds by pairing the spins of these electrons with unpaired electrons of four other atoms. The simplest example is the formation of four bonds to four hydrogen atoms in the methane molecule, CH4. The energy put in to promote the 2s electron is more than made up for by the formation of two extra C-H bonds, that would not have otherwise been possible.
The picture is still incomplete. The bonding picture in CH4 implies that we have three σ-bonds of one type, by pairing three of the H1s with the C2p electrons, and one σ-bond of another type from pairing the remaining H1s and C2s electrons. However, we know experimentally that all four C-H bonds in the methane molecule are identical, having the same bond strengths and length. This discrepancy is overcome because quantum mechanics allows us to describe the same electron density in a variety of equivalent ways. With the excited carbon atom, we can consider the electron distribution as being derived from having four unpaired electron in a 2s orbital and three 2p orbitals. We can also describe it, however, as arising from having the four unpaired electrons in four equivalent orbitals that result from mixing the original atomic orbitals. Mixed orbitals derived from the atomic orbitals on the same atom are termed hybrid orbitals and are formed by the interference between the wave functions of the individual atomic orbitals. The combinations that give the four hybrid orbitals for the carbon atom in methane are described mathematically as:
h1 = s + px + py + pz
h2 = s - px - py + pz
h3 = s - px + py - pz
h4 = s + px - py - pz
What does this actually look like? The interference between the positive and negative parts of the wave functions for the s and p-orbitals results in four hybrid orbitals with large lobes pointing towards the corners of a regular tetrahedron centred on the nucleus of the carbon atom. As these hybrids are constructed from one s and three p orbitals, they are called sp3 hybrid orbitals. Using the concepts of promotion and hybridisation we can now rationalise the tetravalent nature of carbon and the equivalency of the four C-H bonds in the methane molecule.
We also have molecules containing carbon atoms which are bonded to only three atoms, for example ethene H2CCH2. This is a planar molecule with HCH and HCC bond angles of 120°. We still consider the carbon atoms in this molecule to be promoted, having the electronic structure 2s12p3 but here we form three sp2 hybrid orbitals using the s orbital and two of the p orbitals. Expressed mathematically the combinations for these are:
h1 = s + √2px
h2 = s + √(3/22px) - √(1/32py)
h3 = s - √(3/22px) - √(1/32py)
The three sp2 hybrid orbitals lie in the same plane, pointing towards the corners of an equilateral triangle. Each carbon atom in ethene then forms two σ-bonds to hydrogen and forms a linking C-C σ-bond. The remaining 2pz orbitals which are not involved in the hybridisation lie perpendicular to this plane. The unpaired 2pz electrons of the two CH2 fragments can pair up by merging sideways forming a π-bond, just as we saw for N2. The resulting C=C double bond locks the molecule into a planar arrangement as any rotation about the C-C bond weakens the π-bond, leading to an overall increase in the energy of the molecule.
This can easily be extended to the ethyne, HCCH in which each carbon only bonds to two atoms. Here we form two sp hybrid orbitals on carbon using the 2s and 2pz orbitals with the combinations:
h1 = 2s + 2pz
h2 = 2s - 2pz
The large lobes of the two hybrid orbitals of the carbon atoms are at 180° to each other, giving a linear molecule. One hybrid orbital forms a σ-bond to a hydrogen atom while the other forms a σ-bond to the other carbon. The remaining unpaired electrons in the 2px and 2py orbitals of the carbon atoms can then pair to form two π-bonds resulting in a C≡C triple bond.
So in VB theory, we have N atomic orbitals and hence N hybrid orbitals giving us N valence bond wave functions. Compounds of the second row of the periodic table, boron to neon, obey what is called the 'octet rule'. This states that the valence shell of an atom, through bonding interactions, will contain eight, or an octet, of electrons. This arises as we form bonds through the four available valence atomic orbitals, the 2s and three 2p. Each of these orbitals, whether engaging in bonding or not, can only accommodate a pair of electrons. Using heavier atoms of third row elements and below, we can form more complicated molecules where the octet rule is exceeded and there are more than eight electrons in the valence shell. The d-orbitals are now in the valence shell of these atoms and so more exotic hybrid orbitals can be formed through combinations of s, p and d orbitals allowing the accommodation of more electron pairs.
| Number of hybrid orbitals | Shape | Hybridisation |
|---|---|---|
| 2 | Linear | sp |
| 3 | Trigonal planar | sp2 |
| 4 | Tetrahedral | sp3 |
| 5 | Trigonal bipyramidal | sp3d |
| 6 | Octahedral | sp3d2 |
The hybridisation described in the table involves the 'pure' involvement whole numbers of atomic orbitals, however it is possible to only use intermediate proportions of atomic orbitals in the hybridisation. We can see that as the p character of the sp-hybridised orbitals increases from sp (180°) through sp2 (120°) to sp3 (109°), the angle between these orbitals gets closer to the angle between individual p orbitals (90°). Therefore, for water, where we know the bond angle to be 104°, we can say that the orbitals that are forming bonds to hydrogen are somewhere between pure p orbitals and sp3 hybrid orbitals.
Resonance
Resonance is a special feature of molecular structure that comes out of VB theory and involves superimposing different electron density distributions on the same molecular framework. In the molecule HCl we might consider the bonding picture to be entirely covalent and so the wave function would be written as:
ψcov = ψH1s(1)ψCl2pz(2) + ψH1s(2)ψCl2pz(1)
The bond is formed by the pairing of the unpaired electrons in the H1s and Cl2pz orbitals. This description allows one of the electrons in HCl to be on H or on Cl and allows both electrons to be only on one atom at the same time. However, Cl is much more electronegative4 than H and so we would expect that the electrons in the bond would be pulled more towards the Cl atom. So we would expect to get a contribution to the overall electron distribution from that of the polar molecule H+Cl- whose wave function would be:
ψion = ψCl2pz(1)ψCl2pz(2)
As we expect the overall electron distribution to be somewhere between the two possibilities, we can superimpose their wave functions to give:
ψ = ψcov + λψion
where λ is a coefficient that describes the weighting of the ionic wave function to the overall wave function. The value of λ can be determined by varying it and solving the Schrodinger Equation for each value until an energy minimum is reached. This overall wave function is a mixture of the covalent and ionic wave function and is called a resonance hybrid.
This approach can also be extended to other types of molecule, for example benzene. This molecule is constructed from six sp2 hybridised carbon atoms, each bonded to one hydrogen atom. The six CH fragments form a six membered ring, with each carbon atom possessing an unpaired electron in it's pz orbital available to form a total of three π-bonds. The Kekule structure of benzene is drawn with three C=C double bonds with C-C single bonds between them, and we would expect that the single bonds, being weaker, should be longer than the double bonds. However, we know that all the C-C bonds in benzene are of the same length and strength. If we number the carbon atoms 1 to 6 and try to draw in where the double bonds should be, we can draw them C1 to C2, C3 to C4 and C5 to C6, which we will call Kekule structure 1. But we can also draw the equally likely possibility called Kekule structure 2 where the double bonds are C2 to C3, C4 to C5 and C6 to C1. Since both are equivalent we can superposition their individual wave functions to give an overall wave function combining the two Kekule structures:
ψ = ψKek1 + ψKek2
This makes all the C-C bonds equivalent as we determined experimentally. The energy of the resultant wave function is lower than that of the two possible Kekule structures and this is known as resonance stabilisation. This explains the stability of benzene and its relative inertness, generally requiring harsh conditions to react.
Conclusions
Valence bond theory provides an adequate rationalisation for many aspects of molecular structure and bonding through the reduction in energy on the pairing of the unpaired electrons of constituent atoms. It can be used to successfully predict the structures and properties or molecules and their probable reactivity. The more accurate and developed theory of chemical bonding that is used in virtually all modern computational work is molecular orbital theory, described in this entry.
References
- Atkins, P.W., Physical Chemistry, 6th Edn, OUP